5. Future features
cookbook.RmdThese are rough implementations of future features. They are intended
to extend bubblers base functionality using various
packages. If you want to help implement these I will accept PRs.
Technique 1: Adding a bray-curtis tree to a bar plot
library(bubbler)
library(phyloseq)
# library(tidyverse)
library(dplyr)
library(tibble)
library(tidyr)
library(ggplot2)
library(vegan)
library(ape)
library(patchwork)
library(viridis)
# library(ggtree) loading ggtree causes namespace issues
library(ggnewscale)
# otufile = system.file("extdata", "gp_otu_table_rand_short.txt.gz", package="phyloseq")
# mapfile = system.file("extdata", "master_map.txt", package="phyloseq")
# trefile = system.file("extdata", "gp_tree_rand_short.newick.gz", package="phyloseq")
# rs_file = system.file("extdata", "qiime500-refseq.fasta", package="phyloseq")
# qiimedata = import_qiime(otufile, mapfile, trefile, rs_file, verbose = FALSE)
counts_q <- system.file("extdata", "qiime", "table-dada2.qza", package = "bubbler")
taxa_q <- system.file("extdata", "qiime", "taxonomy.qza", package = "bubbler")
metadata_q <- system.file("extdata", "qiime", "sample-metadata.tsv", package = "bubbler")
# Import the data compute bray-curtis dissimilarity matrix and cluster
# asv <- data.frame(phyloseq::otu_table(qiimedata))
asv <- asv_data_qiime(counts_q)
asv <- asv %>%
as.data.frame() %>%
column_to_rownames(var = "sample_id") %>%
as.matrix()
bc_dist <- vegan::vegdist(asv, method = "bray")
hc <- hclust(bc_dist, method = "average")
# Have to convert from a phylo to a ggtree object to get at the tip order.
phylo_tree <- as.phylo(hc)
# ggtree_plot <- ggtree::ggtree(phylo_tree)
tree_data <- ggtree::fortify(phylo_tree)
tree_data <- tree_data %>%
left_join(meta_data_qiime(metadata_q), by = c("label" = "sample_id"))
ggtree_plot <- ggtree::ggtree(tree_data) +
ggtree::geom_tippoint(aes(color = body_site)) +
scale_color_viridis_d()
# Get the tip order.
tip_order <- ggtree_plot$data %>%
filter(isTip == TRUE) %>%
arrange(y) %>%
select(label) %>%
rev() %>%
pull()
# Make the relative abundance table and the sideways barplot.
q <- rel_abund_qiime(counts_q, taxa_q, metadata_q) %>%
pool_taxa(n_taxa = 12, keep_metadata = TRUE)
p2 <- q %>%
arrange_var(levels = tip_order) %>%
bar_plot(position = "fill") +
labs(x = NULL) +
scale_x_discrete(position = "top") +
scale_y_continuous(expand = c(0,0)) +
coord_flip() +
theme(plot.margin = margin(0, 0, 0, 5))
ggtree_plot + p2 + patchwork::plot_layout(guides = "collect")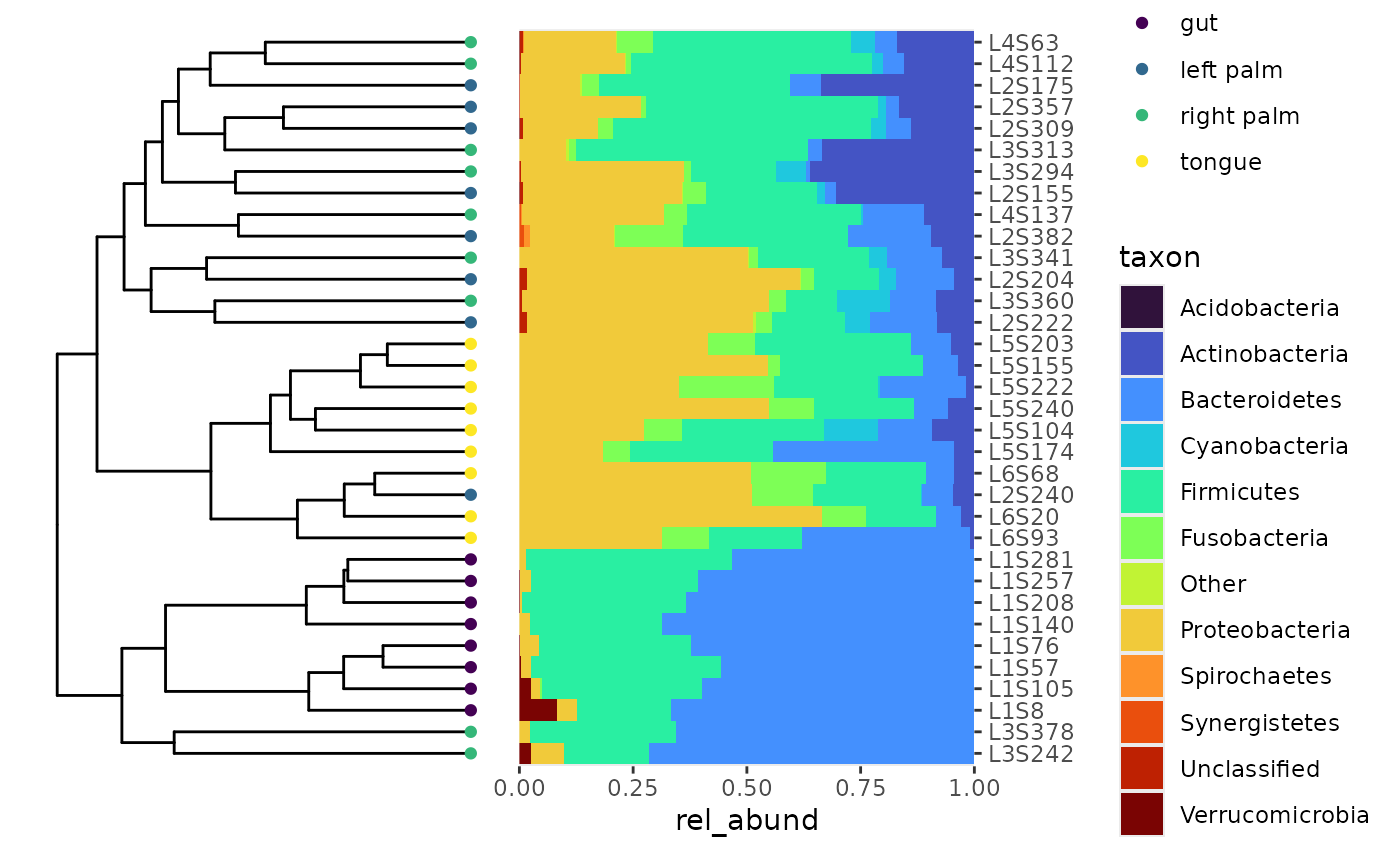
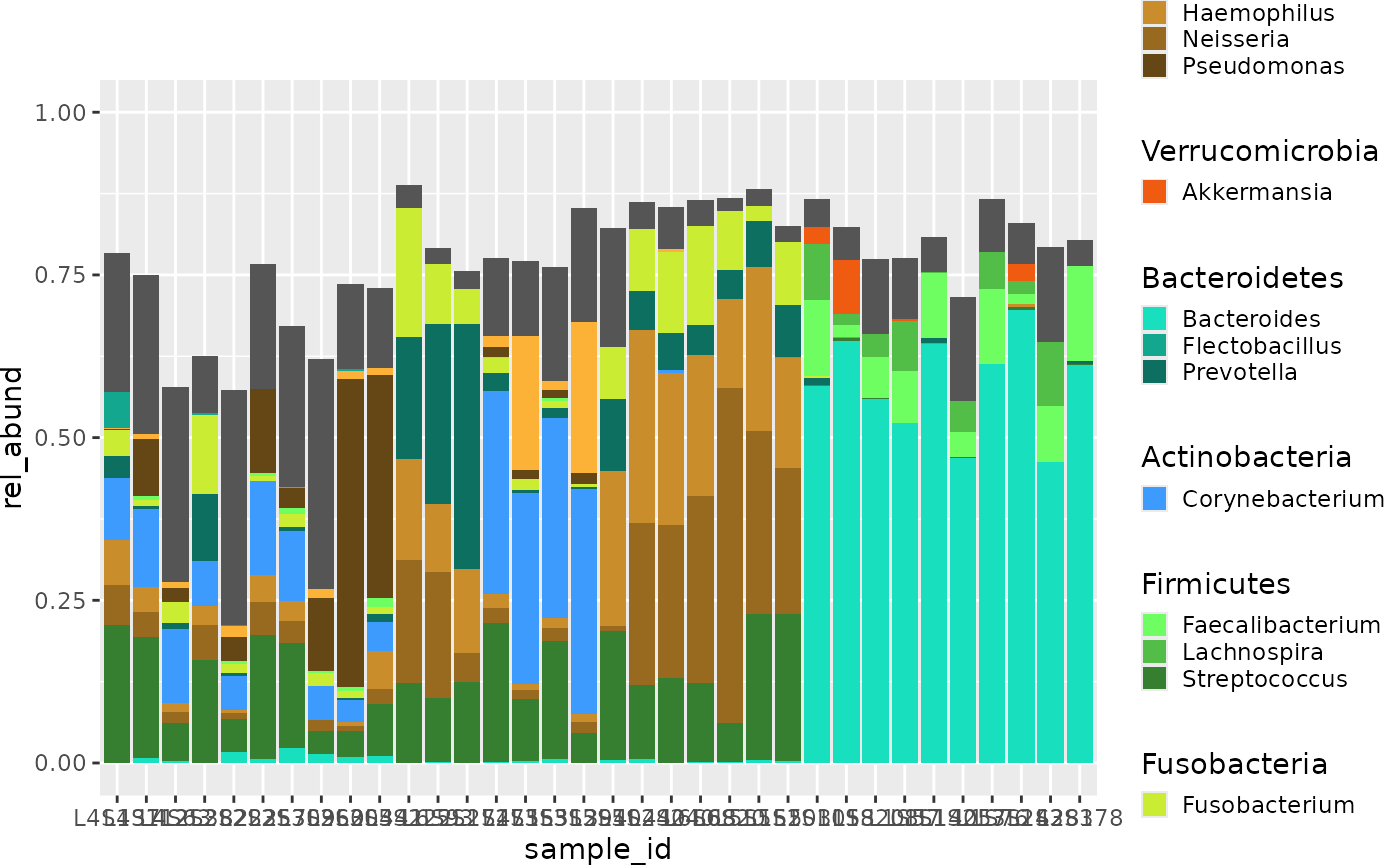
Technique 2: Nested legend and colourscheme within taxa
###~%~ trying to get a nested colour scheme for phylum ~%~###
taxa_qiime <- system.file("extdata", "qiime", "taxonomy.qza", package = "bubbler")
tx <- taxa_data_qiime(taxa_qiime)
asv_qiime <- system.file("extdata", "qiime", "table-dada2.qza", package = "bubbler")
taxa_qiime <- system.file("extdata", "qiime", "taxonomy.qza", package = "bubbler")
metadata_qiime <- system.file("extdata", "qiime", "sample-metadata.tsv", package = "bubbler")
# generate rel_abund
q <- rel_abund_qiime(
asv_qiime = asv_qiime,
taxa_qiime = taxa_qiime,
metadata_qiime = metadata_qiime,
taxa_level = "Genus", )
# pool taxa, rename taxon to Genus (its true level) and select it.
qp <- pool_taxa(q, 0.0035, label = FALSE)
taxa_pooled <- qp %>%
rename(Genus = "taxon") %>%
select(Genus) %>%
distinct()
# merge the filtered
taxa_full <- inner_join(tx, taxa_pooled, by = "Genus" )
phylum_genus <- taxa_full %>%
select(Phylum, Genus) %>%
arrange(Phylum, Genus ) %>%
distinct()
pg_nest <- phylum_genus %>%
group_by(Phylum) %>%
nest()
base_colours <- turbo(n = nrow(pg_nest), begin = 0.2, end = 0.8)
# cutoff <- ceiling(n_rows * 0.7)
2 * 0.8
#> [1] 1.6
# taxon is Genus actually.
tb <- tibble(taxon = character(), color = character())
for(i in 1:nrow(pg_nest)){
phylum <- pg_nest$Phylum[i]
genera <- pg_nest$data[[i]]
phylum_shades_pal <- colorRampPalette(c(base_colours[i], "black"))
cutoff <- ceiling(nrow(genera) * 0.5)
phylum_shades <- phylum_shades_pal(nrow(genera) + cutoff)
phylum_shades <- phylum_shades[1:(length(phylum_shades) - cutoff)]
pgc <- tibble(taxon = pull(genera),
color = phylum_shades)
tb <- bind_rows(tb, pgc)
}
qpc <- left_join(qp, tb, by = "taxon")
idx <- grepl("^<", qpc$taxon)
qpc[idx,"color"] <- "#999999"
qpc[qpc[["taxon"]] == "Unclassified", "color"] <- "#555555"
names(qpc$color) <- qpc$taxon
# colours are now set, now time to group by phylum for plotting!
cols <- qpc$color
tx2 <- tx %>%
select(Genus, Phylum) %>%
rename(taxon = "Genus") %>%
na.omit() %>%
distinct()
qpc2 <- left_join(qpc, tx2, by = "taxon")
idx <- grepl("^<", qpc2$taxon)
qpc2[idx,"Phylum"] <- "Other"
qpc2[qpc2[["taxon"]] == "Unclassified", "Phylum"] <- "Other"
groups <- qpc2 %>%
distinct(Phylum, taxon) %>%
mutate(order = as.numeric(forcats::fct_inorder(Phylum))) %>%
split(.$Phylum)
qpc %>%
arrange_taxa(pooled = "top") %>%
arrange_sample_by_taxa() %>%
ggplot() +
lapply(groups, function(x){
list(
geom_col(aes(x = sample_id, y = rel_abund, fill = taxon), position = "fill"),
scale_fill_manual(name = unique(x$Phylum),
values = cols, limits = x$taxon, na.value = "transparent",
guide = guide_legend(order = unique(x$order))),
new_scale_fill()
)
}) + guides(fill=guide_legend(ncol=3)) +
theme(
# legend.position = "bottom"
plot.margin=margin(t=30),
legend.key.size = unit(10, "pt"),
# legend.position = "bottom"
# legend.box = "vertical"
)